

Does hyperoxia suppress a COPD patient’s respiratory drive? Does it cause V/Q mismatching? Does it change the chemistry of the patient’s blood through the Haldane effect? It’s enough to make you want to give up and page respiratory therapy. Well lucky for you we sifted through the primary literature to bring you the myths and facts, and the short answer is…it’s complicated.
Theory 1: Oxygen Induced Hypoventilation
One commonly cited theory goes like this:
There are two central drivers of respiratory drive, hypercarbia and hypoxemia. Because COPD patients spend their lives chronically hypercarbic they no longer respond to that stimulus, and their only trigger for respiratory drive is the level of oxygen (or lack their of) in their blood. Supplemental O2 removes a COPD patient’s hypoxic respiratory drive causing hypoventilation with resultant hypercarbia, apnea, and ultimate respiratory failure. The first study to really investigate this theory was done in 1980 [1].
Aubier M et al. Effects of the administration of O2 on ventilation and blood gases in patients with chronic obstructive pulmonary disease during acute respiratory failure. Am Rev Respir Dis.1980;122(5):747–754.
Population: 22 ICU patients with known COPD in acute respiratory failure
Methods: Minute ventilation and PaCO2 were measured in all patients first while breathing room air and then while breathing supplemental oxygen.
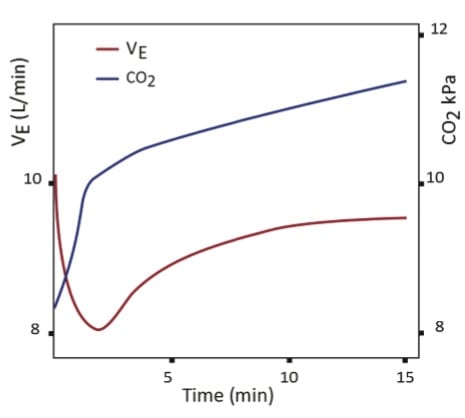
Results: All patients had an initial drop in minute ventilation (Ve) once placed on supplemental O2 but Ve then recovered to near baseline levels. At the same time PaCO2 continued to increase (figure 1). There was NO correlation between minute ventilation and the increase in PaCO2.
Conclusions: The study subjects had a transient drop off in minute ventilation on supplemental oxygen therapy, but it did not correlate with steadily increasing PaCO2 levels. A follow up study showed that respiratory drive and minute ventilation both stayed within normal limits on supplemental O2 [2]. The authors suggested that hypoventilation due to loss of hypoxic respiratory drive WAS NOT the cause of hypercarbia after O2 administration, and that other factors, like the Haldane effect and V/Q mismatching were likely to blame.
Theory 2: The Haldane Effect
This brings us to a second theory, the Haldane effect.
The amine groups of proteins in our blood, like hemoglobin (Hb), combine with CO2 to form carbamino compounds. The ability of deoxygenated Hb to bind CO2 is much higher than that of oxygenated Hb (this makes some physiologic sense, because Pa CO2 should be higher in venous blood). Supplemental O2 shifts the equilibrium between deoxygenated and oxygenated Hb more towards the oxygenated form. This reduces the amount of CO2 that can be bound, and that CO2 winds up dissolved in the blood, resulting in an increased Pa CO2.
The chemistry behind this theory is sound, but it’s proved understandably tricky to study in practice. Again we rely on the 1980 Aubier et al study [1]. They calculated how much of the observed change Pa CO2 could be due to the Haldane effect based on the observed change in Hb O2 saturation in their patients. They concluded that the effect, while presumably real, could only have accounted for about 25% of the increase. In other words there just wasn’t enough bound CO2 to dislodge.
It follows that there must be ANOTHER physiologic change at work, which leads us to…
Theory 3: Ventilation Perfusion (V/Q) Mismatch
The pulmonary vasculature can dilate and constrict to alter blood flow and match ventilation to perfusion (figure 2), and the primary driver of vascular dilation and increased perfusion is alveolar O2.
COPD patients have diseased lungs, and over time their bodies have carefully allocated perfusion to parts of their lungs that work, and away form the parts that don’t. Administering supplemental O2 screws up this careful balance. Diseased sections of lung see increased PaO2 and steal perfusion away from better functioning areas. This results in shunting, dead space ventilation, and eventually hypercarbia.
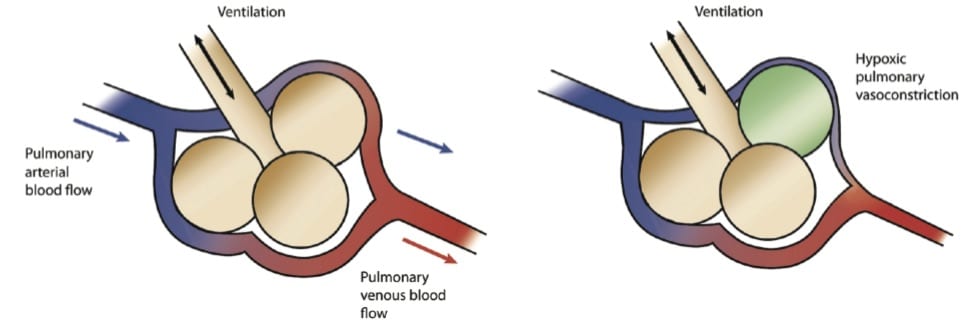
Aubier et al inferred that V/Q mismatch was the primary driver of hypercabia after O2 supplementation, but a follow up study 20 years later looked at it more directly, and their findings muddied the waters [3].
Robinson et al. The role of hypoventilation and ventilation-perfusion redistribution in oxygen-induced hypercapnia during acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161(5):1524–1529.
Population: 22 hospitalized patients with a diagnosis of acute COPD exacerbation
Methods: Inert gas was infused into each patient using a peripheral venous catheter. Expired air makeup and arterial blood gas readings were recorded in each patient while breathing room air, and then 100% supplemental O2. The patients were classified as CO2 ‘retainers’ if their Pa CO2 rose by more than 3mmhg while on supplemental O2 and as ‘non-retainers’ if it did not. The authors then compared minute ventilation, V/Q matching (calculated based on the expired inert gas), and dead space ventilation between the two groups.
Results: Ventilation perfusion heterogeneity (i.e. V/Q mismatch) increased significantly in both the retainer and non-retainer groups. Minute ventilation decreased significantly in the retainers but not the non-retainers. Finally, dead space ventilation increased significantly in the retainer group, but was unchanged in the non-retainer group.
Conclusions: Since V/Q mismatch increased in both retainers and non-retainers the authors concluded that V/Q mismatch could not be the cause of observed CO2 retention (hypercarbia). Further, because minute ventilation decreased in the retainer group, but remained stable in the non-retainers they proposed that OXYGEN INDUCED HYPOVENTILATION was to blame.
WAIT WHAATT??!!!
That’s right. Robinson et al came to the exact opposite conclusion that Aubier et al did 20 years earlier in their landmark study. They concluded that hypoventilation (theory #1, which I just called a myth) was the cause of oxygen induced hypercapnia in COPD patients. But just in case you’re not confused enough; it’s not quite that simple.
Controversy: Other authors have drawn different conclusions from the Robinson et al study. For example, Abdo and Heunks [4] suggest that hypercarbia in the retainer group was likely due to the increase in dead space ventilation (which was unchanged in the non-retainers). The retainer group did show a decrease in minute ventilation, but so did the patients in Aubier’s 1980 study (that decreases just wasn’t large enough to account for all the observed hypercarbia). It’s plausible that although V/Q mismatch increased in both groups, it only caused hypercarbia in the retainer group because it increased dead space ventilation in their lungs.
It’s an interesting theory and it fits nicely with the prior literature, but it’s important to stress that it directly contradicts the authors who actually conducted the study.
The Bottom Line:
I hope this has been an enjoyable tour through pulmonary physiology. Let’s see if we can make any sense of the myths and facts of oxygen induced hypercarbia in COPD patients.
Theory 1: Oxygen Induced Hypoventilation
Myth or Fact?
Unclear
Both large studies that looked at this theory found that supplemental O2 does lead to hypoventilation. Aubier et al. concluded that it was transient and DID NOT account for the observed hypercarbia. Robinson et al. concluded that IT DID.
Theory 2: The Haldane Effect
Myth or Fact?
Probably Fact… But
The chemistry is sound, and the effect likely causes some hypercarbia, but we have minimal direct evidence for it. It also cannot account for all of the hypercarbia that has been observed in the above studies.
Theory 3: V/Q Mismatch
Myth or Fact?
Unclear
Aubrier et al. concluded by process of elimination that V/Q mismatch should be responsible for most of the observed hypercarbia. Robinson et el found that V/Q mismatch increased in both retainers and non-retainers and therefore could not be the cause; however, other authors point out that dead space ventilation increased in the retainer group, and suggest that this specific type of V/Q mismatch could be the cause of hypercarbia.
Guest Contributor:
 Allan Guiney, MD
Allan Guiney, MD
Resident Physician
NYU/Bellevue Department of Emergency Medicine
New York, NY
References
- Aubier M, Murciano D, Milic-Emili J, et al. Effects of the administration of O2 on ventilation and blood gases in patients with chronic obstructive pulmonary disease during acute respiratory failure. Am Rev Respir Dis.1980;122(5):747–754. PMID: 66778278
- Aubier M, Murciano D, Fournier M, Milic-Emili J, Pariente R, Derenne JP: Central respiratory drive in acute respiratory failure of patients with chronic obstructive pulmonary disease. Am Rev Respir Dis 1980, 122:191-199. PMID: 67746339
- Robinson TD, Freiberg DB, Regnis JA, Young IH. The role of hypoventilation and ventilation-perfusion redistribution in oxygen-induced hypercapnia during acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161(5):1524–1529. PMID: 10806149
- Abdo WF, Heunks LMA: Oxygen-induced hypercapnia in COPD: myths and facts. Critical Care 2012, 16:323. PMID: 23106947
Post Peer Reviewed By: Salim Rezaie (Twitter: @srrezaie)

